In the conventional ADC narrative, the antibody picks the cell, the payload kills it, and the linker just holds them together until they get there. The picture is wrong in roughly the same way that calling a fuel injector “plumbing” is wrong.
The linker is what decides whether your ADC actually works.
It is also the part of an ADC most underestimated by teams coming from small-molecule programs. The antibody side is solved chemistry — by which we mean it is biology that biology teams know how to handle. The payload, increasingly, is reused: most pipeline ADCs carry an MMAE, MMAF, DM1, DM4, SN-38, or DXd analog. The variable that distinguishes Adcetris from Kadcyla from Enhertu — three vedotin-, maytansinoid-, and exatecan-bearing ADCs that all reached approval — is what sits between the antibody and the drug. Stability profile, release kinetics, bystander effect, DAR distribution, aggregation propensity. The linker is the differentiating molecule.
This post is a quick field guide to the chemistry that lives between the mAb and the warhead. We keep biology to the minimum required to make the chemistry decisions intelligible.
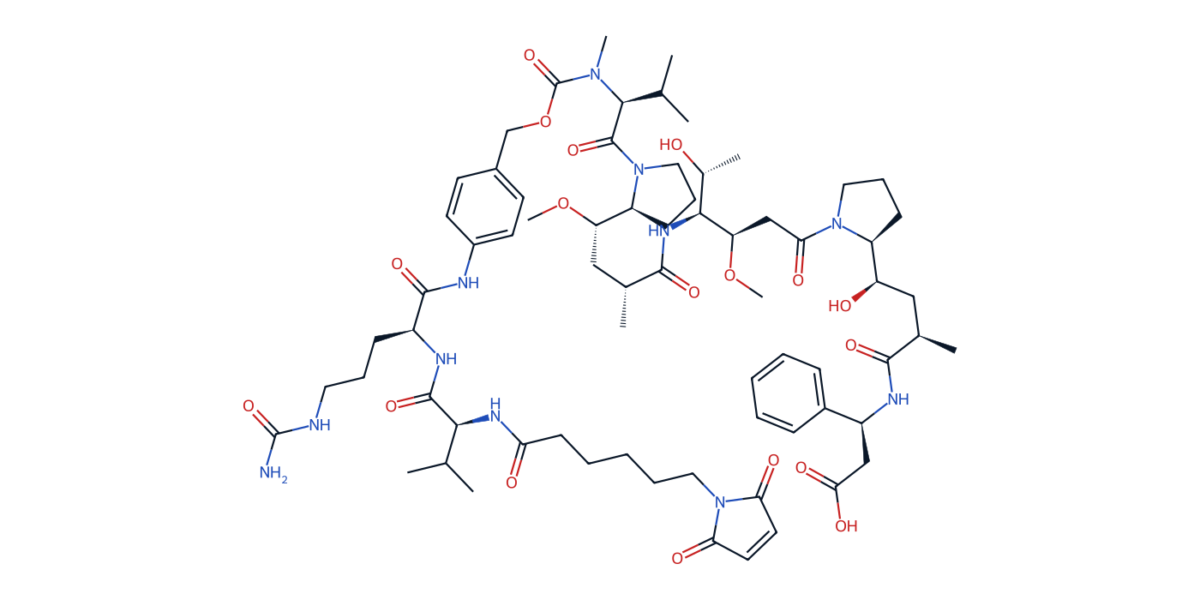
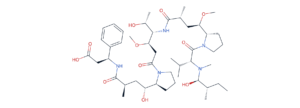
MC-vc-PAB-MMAE — the linker-payload from brentuximab vedotin (Adcetris). Maleimide for cysteine conjugation (bottom right), caproyl spacer, Val-Cit dipeptide, para-aminobenzyloxycarbonyl (PABC) self-immolative spacer, and the MMAE payload (upper right). Every region of this molecule is a separate design decision.
The two big linker families
Linkers are conventionally divided into cleavable and non-cleavable.
A cleavable linker is one that, once the ADC has internalized into the target cell, breaks open under a specific intracellular trigger and releases the payload (or a payload-linker fragment) as a small molecule. Cleavable linkers split into four chemistry classes by what triggers the cleavage:
• Protease-cleavable (peptide-based). The dominant chemistry. A short peptide — most commonly Val-Cit, sometimes Val-Ala or Gly-Gly-Phe-Gly — is recognized by the lysosomal cysteine protease cathepsin B, which cuts the C-terminal amide. A para-aminobenzyloxycarbonyl (PABC) self-immolative spacer then releases the free payload. This is the chemistry behind Adcetris, Polivy, and Padcev (Val-Cit-PABC) and Enhertu (the four-residue Gly-Gly-Phe-Gly tail with an aminomethyl spacer).
• Acid-labile (hydrazone). Hydrolyzes in the acidic lysosomal compartment. Used in the first-generation Mylotarg and still present in a few clinical-stage programs. Premature release at pH 7.4 in plasma is the main liability.
• Reducible (disulfide). Reduced by intracellular glutathione, which sits in the millimolar range inside cells versus micromolar in plasma. Used with maytansinoid payloads — DM4 in mirvetuximab soravtansine (Elahere) is the canonical example. Hindered disulfides (one or two methyl substituents adjacent to sulfur) tune the reduction kinetics.
• Glycosidase-cleavable (β-glucuronide). Newer; cleaved by lysosomal β-glucuronidase. Hydrophilic, which helps with aggregation; appears in several Phase 1/2 programs.

The Val-Cit-PABC linker. The cathepsin B cleavage site is highlighted in red — the amide bond between Cit C(=O) and the PABC aniline N. Once cut, the PABC group self-immolates by 1,6-elimination, ejecting CO₂ and releasing the free payload.
A non-cleavable linker is one where the drug is liberated only after the antibody itself has been proteolytically destroyed in the lysosome. What gets released is a small-molecule-linker-amino-acid adduct, often with the linker still attached to a lysine or cysteine residue. The classic example is SMCC in T-DM1 (Kadcyla) — succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate — which releases lysine-MCC-DM1 as the active species. Non-cleavable linkers give the cleanest plasma profile but no bystander effect: the released payload is too polar to leave the cell.

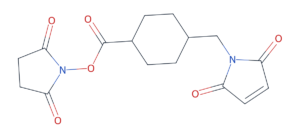
SMCC — the non-cleavable linker used in Kadcyla (T-DM1). The NHS active ester (left) acylates antibody lysines; the maleimide (right) conjugates to the maytansinoid payload’s free thiol. The drug is only released after the entire antibody is digested.
The right linker chemistry depends on the payload, the target’s expression pattern, the trafficking biology, and the tolerability profile you can live with. There is no winner across all parameters.
Three chemistry problems the linker has to solve
1. Stability versus release — the central tradeoff
The fundamental problem is that the linker has to be perfectly stable in circulation for a week and then cleave with high efficiency in a lysosome within hours. Any drift toward the unstable end produces premature payload release in plasma — which is toxicity without targeting. Any drift toward the stable end means most internalized ADC is recycled before its payload escapes, which is delivered drug that does no work.
The most-studied failure mode is the maleimide retro-Michael. The thioether bond formed between a maleimide and an antibody cysteine is reversible. The thiosuccinimide can re-eject its thiol — slowly in vitro, faster in the presence of plasma albumin, which has a free cysteine of its own that can pull the payload-linker off the antibody and walk away with it. The classic fix is succinimide hydrolysis — ring-opening the succinimide to a stable thioether-substituted succinamide. Self-stabilizing maleimides, with electron-withdrawing groups that drive on-conjugation hydrolysis, are now a standard solution. Newer chemistries — pyridazinedione, vinyl sulfone, dibromopyridazinedione — avoid the problem by being non-reversible from the start.

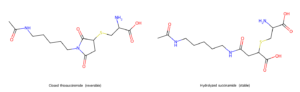
The maleimide retro-Michael fix. Left: the closed thiosuccinimide formed by cysteine addition to a maleimide — reversible, can release the payload back into plasma. Right: the hydrolyzed succinamide, ring opened to a free carboxylic acid — no longer reversible, locked in.
2. The conjugation site decides the homogeneity story
Conjugation chemistry determines what “DAR 4” actually means in your vials. With random cysteine conjugation — the standard approach for Adcetris, Enhertu, Polivy — you reduce the four interchain disulfides of an IgG to liberate eight thiols, then run a Michael addition with maleimide-linker-payload. The result is a Poisson-distributed mixture of DAR 0, 2, 4, 6, 8 species in known ratios. You can shift the distribution by titrating reducing agent but you cannot make it monodisperse.
Random lysine conjugation (used for Kadcyla) gives even broader heterogeneity — IgG has roughly eighty surface-accessible lysines and you typically modify two to five. Both the DAR distribution and the positional distribution matter, and you cannot easily separate the two by chromatography.
Site-specific conjugation is the modern answer: engineer specific cysteines into the antibody (Genentech’s THIOMAB platform), incorporate unnatural amino acids (pAcF, azidophenylalanine), or use enzymatic tags (sortase, transglutaminase, glycoengineering). The result is a near-monodisperse DAR 2 or DAR 4 species, and the site itself becomes an optimizable variable. Programs in clinical development increasingly use site-specific approaches — partly for the cleaner PK, partly because reduced heterogeneity removes a category of process and regulatory headaches downstream.
The bench cost is real. Site-specific conjugation requires engineered antibodies, characterization of every new site, and orthogonal linker chemistry. Random cysteine remains the default for first-in-class programs because it is fast and well-understood.
3. Hydrophobicity is a hidden ADC liability
The cytotoxic payloads of choice — MMAE, DM1, calicheamicin, PBD dimers — are aggressively lipophilic. Loading multiple copies of one of these onto an antibody without aggregation is a chemistry problem the linker has to help solve.

MMAE — monomethyl auristatin E. Five peptide bonds, four stereocenters, two methoxy groups, multiple methylated nitrogens. Highly lipophilic by design (it has to cross cell membranes after release) and a significant aggregation driver when loaded at high DAR.
The standard mitigation is to make the linker itself hydrophilic. PEG-based linkers, sulfonate-decorated peptide linkers, and glucuronide linkers all serve this function. Enhertu’s GGFG-aminomethyl spacer, designed at Daiichi Sankyo, is partly chosen for hydrophilicity — it tolerates a high DAR of 8 without aggregation, which is the basis of Enhertu’s clinically useful bystander effect at standard doses.
When this mitigation is missed, you discover it during scale-up. ADCs that pass DAR 4 cleanly may aggregate badly at DAR 8 or higher, fail size-exclusion QC, or precipitate from formulation buffer overnight. The linker design decisions made on the bench at milligram scale propagate downstream in expensive ways.
What this means for outsourced linker chemistry
Two recurring asks from biotech teams bringing ADC linker work to a CRO.
First, the linker construct is its own multi-step synthesis — typically eight to fifteen steps from commercial starting materials, often with three or four orthogonal protecting-group strategies running in parallel for the maleimide, the peptide, the payload’s nucleophilic handle, and the carbamate or carbonate connection to the drug. Programs that scope the linker as “we’ll just have someone make it” routinely underestimate timelines. The linker is its own small-molecule program, and benefits from being treated as one.
Second, the analytical demands are heavier than for a small molecule of similar complexity. Cleavable linker integrity has to be confirmed both before conjugation (as the free linker-payload species) and after (on the intact conjugate). Plasma stability assays, in-vitro lysosomal cleavage assays, and intact-mass LC-MS on the conjugated antibody (or peptide-mapping if site-specific) are part of routine release. Programs that under-invest in this end up surprised by their first PK study.
The frontier
Three places where ADC linker chemistry is moving fastest.
Bispecific ADCs. Dual-payload constructs with two orthogonal linker chemistries releasing different cargo on different triggers. The chemistry is interesting because the two linkers cannot interfere with each other’s conjugation or cleavage, and the antibody scaffold has to accommodate both without aggregation.
Conditionally-stable linkers. Linkers stable in circulation that respond to tumor-microenvironment triggers — low pH, hypoxia-responsive nitroreductase, tumor-specific proteases — rather than ubiquitous lysosomal enzymes. Clinical data is early; the chemistry pipeline is rich.
Immune-modulating payloads. TLR agonists, STING agonists, NLRP3 agonists, RIG-I agonists. The pharmacology is the opposite of cytotoxic chemistry — you want sustained intracellular signaling, not cell death — and the linker chemistry follows. Release in pharmacologically active form, often in a different cellular compartment than for cytotoxics, often at substoichiometric loading.
Each of these moves linker chemistry further from “plumbing” and closer to where small-molecule medicinal chemistry was twenty years ago — a real design discipline with its own SAR.
Up next
Future posts in this series will go deeper into specific corners — Val-Cit-PABC self-immolation mechanism in detail, payload chemistry across cytotoxic classes, site-specific conjugation by enzyme tagging — and on case studies from our own bench.
If your team is working on an ADC linker design or has an existing program where the linker chemistry is the gating step, write to us at info@astinovabiolabs.com.